
Translate this page into:
A rare coexistence of mycosis fungoides and myelodysplastic neoplasm
*Corresponding author: Leny Mathew, Department of Dermatology Venereology and Leprosy, Pushpagiri Institute of Medical Sciences and Research Centre, Thiruvalla, Kerala,India. leny_mathew11@yahoo.com
-
Received: ,
Accepted: ,
How to cite this article: Mathew L, Pappen TT, Kunjukunju BP, Joseph A, Jayaprakash A. A rare coexistence of mycosis fungoides and myelodysplastic neoplasm. J Skin Sex Transm Dis. 2024;6:170-5. doi: 10.25259/JSSTD_5_2024
Abstract
This case report details the presentation and management of a 71-year-old man with a known history of myelodysplastic neoplasm presenting with hypopigmented flat lesions on his chest, abdomen, and extremities over a three-month period, with mild scaling and severe pruritus and evaluated with a detailed dermatological examination and a multidisciplinary diagnostic approach. Laboratory findings showed anemia, leukopenia, and dysgranulopoiesis on peripheral smear. Chromosome analysis revealed an abnormal male complement with Y chromosome loss in 70% of cells. Bone marrow biopsy and aspiration cytology confirmed myelodysplastic syndrome (MDS) with ringed sideroblasts. Skin biopsy showed early-stage mycosis fungoides (MF), positive immunohistochemistry for CD3, CD5, CD8, and Ki-67 index over 70%. Treatment involved weekly blood transfusions, erythropoietin injections, and supportive management for MDS, while MF was managed with topical emollients, antihistamines, and psoralen ultraviolet A therapy. Monthly follow-up demonstrated symptomatic improvement of the skin condition but worsening of general status of the patient. The patient’s unique clinical course highlights the complexity of managing dual malignancies, requiring a tailored and multidisciplinary approach.
Keywords
Mycosis fungoides
Myelodysplastic syndrome
Myelodysplastic neoplasm
Hypopigmented lesions

INTRODUCTION
Mycosis fungoides (MF) is a cutaneous T-cell lymphoma (CTCL) characterized by infiltration of skin with T-lymphocyte-containing patches, plaques, and nodules. The disease’s clinical course and presentation are highly variable.[1] Baron Jean-Louis Alibert, a French dermatologist, coined the name “mycosis fungoides” (1768–1837). Given the mushroom-like appearance of the skin lesions, he proposed that term.[2]
A heterogeneous collection of hematologic neoplasms known as myelodysplastic syndrome (MDS) is defined as a clonal disease of hematopoietic stem cells (HSCs) that result in dysplasia and inefficient bone marrow hemopoiesis. It is possible for some MDS patients to develop acute myeloid leukemia (AML).[3]
CASE REPORT
A 71-year-old man, a known case of myelodysplastic neoplasm since one year duration, came to our outpatient department with complaints of multiple hypopigmented flat lesions over front of chest and abdomen, since three months duration and the lesions gradually progressed in size and number and spread to involve back of trunk and both limbs [Figures 1 and 2].
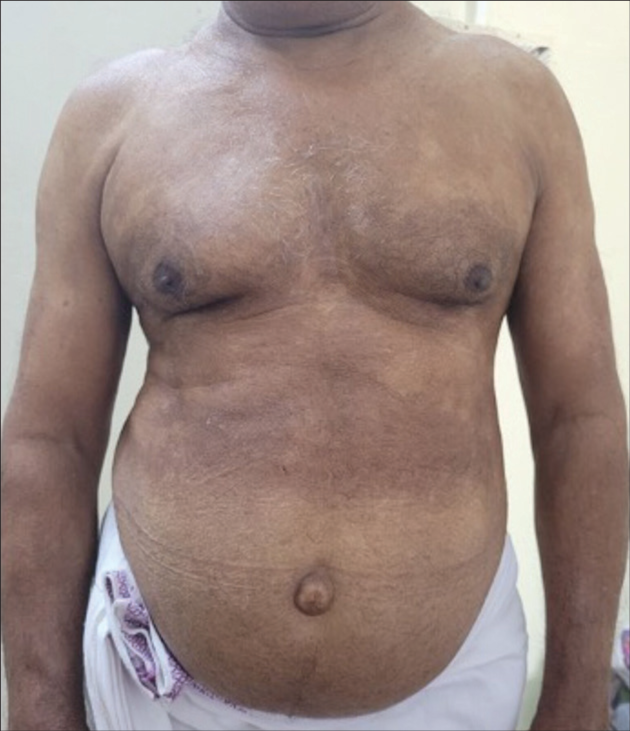
- Multiple discrete and confluent hypopigmented macules with annular borders present over chest, abdomen, and upper limbs.
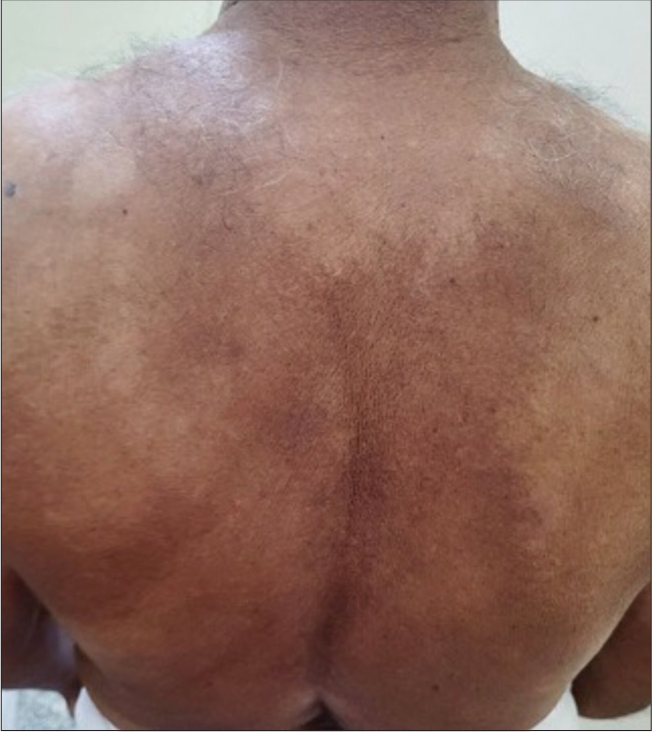
- Multiple discrete and confluent hypopigmented macules with polycyclic borders present over back of trunk with islands of normal skin in between lesional skin.
It was associated with mild scaling and severe itching.
Past history revealed progressive fatigue and shortness of breath of one year duration, recurrent upper respiratory tract infections and intermittent fever since past nine months and had markedly reduced blood counts and was referred to medical oncologist, and on further evaluation, a diagnosis of myelodysplastic neoplasm was made.
There was no history of atopy, bleeding from any sites, loss of appetite, weight loss, recent intake of medications, or recent blood transfusions.
Examination revealed multiple discrete and confluent hypopigmented macules of varying sizes, with polycyclic and annular borders over front and back of chest and abdomen, bilateral upper limbs and lower limbs with areas of normal skin in between.
The lesional surface showed mild fine scaling and atrophy.
Blood hemogram showed hemoglobin – 7.6 g/dL, red blood cell (RBC) count – 2.46 million/mm3, total leukocyte count – 4.07 K/uL, with neutrophils – 8%, lymphocytes – 56%, monocytes –32%, and platelet count – 1.85 K/uL.
Peripheral smear [Figure 3] revealed anisopoikilocytosis, normocytic normochromic RBC with microcytes, tear drop cells, elliptocytes, occasional target cells, occasional spherocytes, few polychromatophils, white blood cell total count of 3900 cells, dysgranulopoiesis in the form of hypolobulation and hypogranulation in 75% neutrophils with few reactive lymphocytes, and no blast cells or atypical cells and normal platelets, thus giving the impression of normocytic normochromic anemia with leukopenia with marked dysgranulopoiesis [Figure 4].
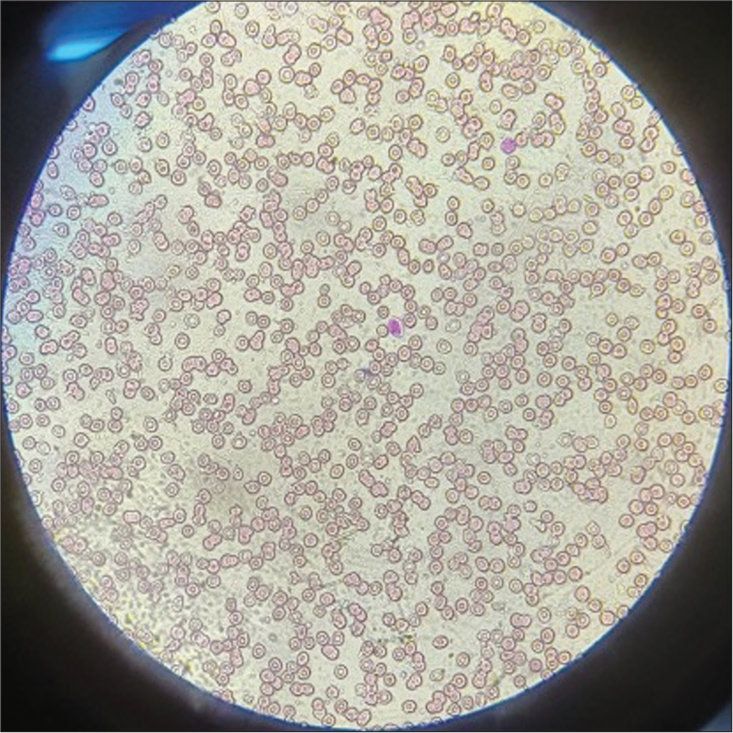
- Peripheral smear showing anisopoikilocytosis, normocytic normochromic red blood cells with microcytes, tear drop cells, elliptocytes, and occasional target cells. Leishman stain, 40×.
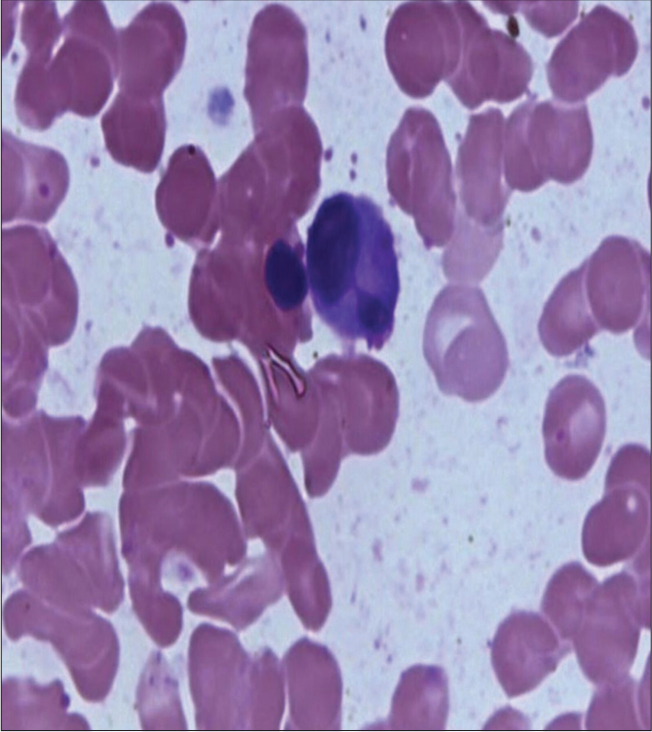
- White blood cell showing hypolobulation. Leishman stain, 100×
Chromosome analysis revealed an abnormal male chromosome complement with loss of Y chromosome in 70% cells.
Bone marrow biopsy shows hypercellular marrow with erythroid hyperplasia and maturing myeloids interspersed in between, with normal megakaryocytes and no atypical cells.
Bone marrow aspiration cytology revealed myelodysplastic syndrome (MDS) with ringed sideroblasts, unilineage dysplasia with cell count as follows: erythroid 53%, promyelocytes and myelocytes 4%, metamyelocytes and band forms 30%, neutrophils 7%, eosinophils 1%, lymphocytes 5%, iron stores 2+ to 3+, and ring sideroblasts up to 20%.
According to the WHO HAEM5, our patient satisfies the criteria of MDS with low blasts and ring sideroblasts (where ring sideroblasts of >15% can substitute for wild type SF3B1 mutation) which comes under the classification of MDS with defining genetic abnormality (as the peripheral smear showed no blasts and bone marrow aspiration cytology revealed ring sideroblasts up to 20%).
Histopathology showed epidermis with mild acanthosis, epidermotropism, dermis with scattered lymphocytic infiltrate, and perivascular lymphocytic infiltrate with coarse fibrosis [Figure 5].
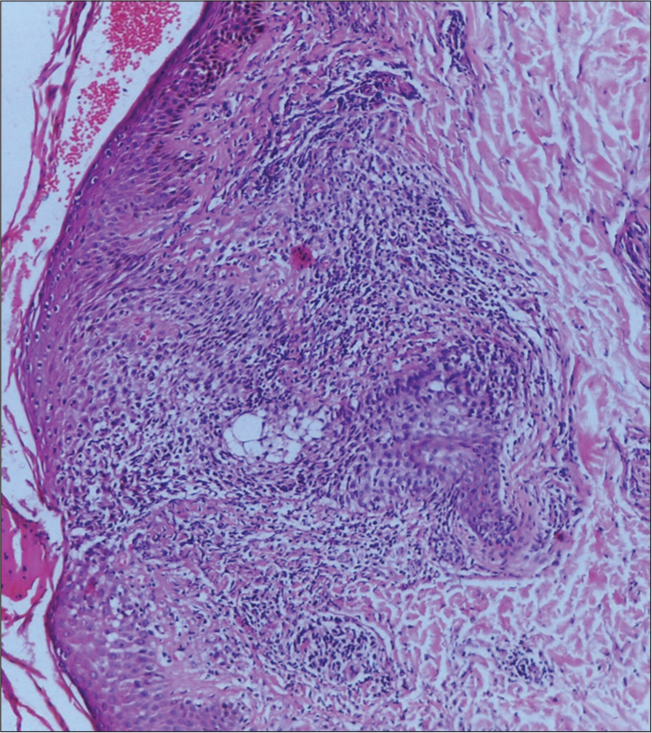
- Histopathology showed epidermis with mild acanthosis, epidermotropism, dermis with scattered lymphocytic infiltrate, perivascular lymphocytic infiltrate with coarse fibrosis. Hematoxylin and Eosin, 10×.
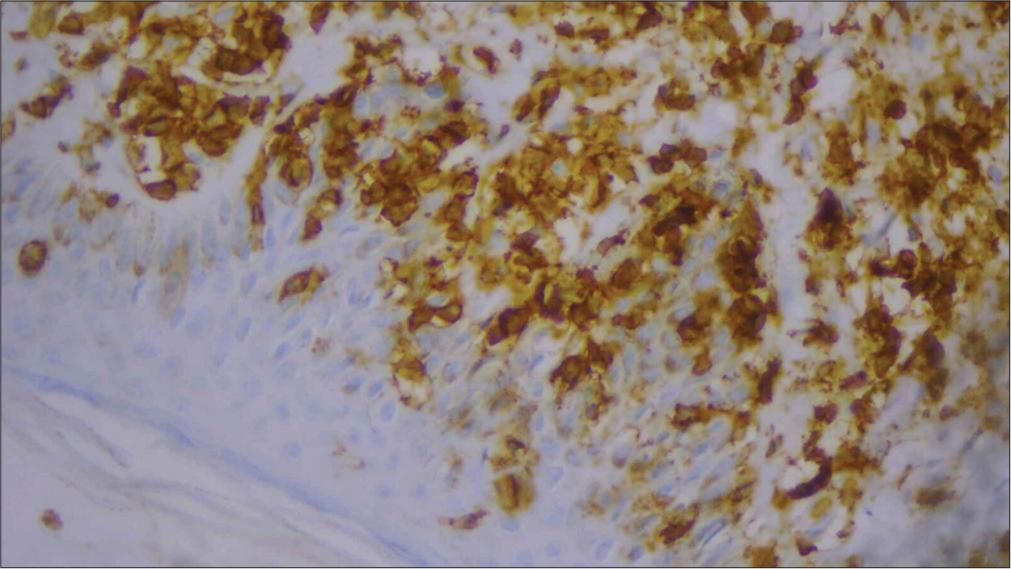
For confirmation, immunohistochemistry (IHC) was done and was positive for CD3, CD5, CD8 [Figures 6-10], CD7 results were negative, and Ki-67 index over 70% was obtained [Figures 11-13].
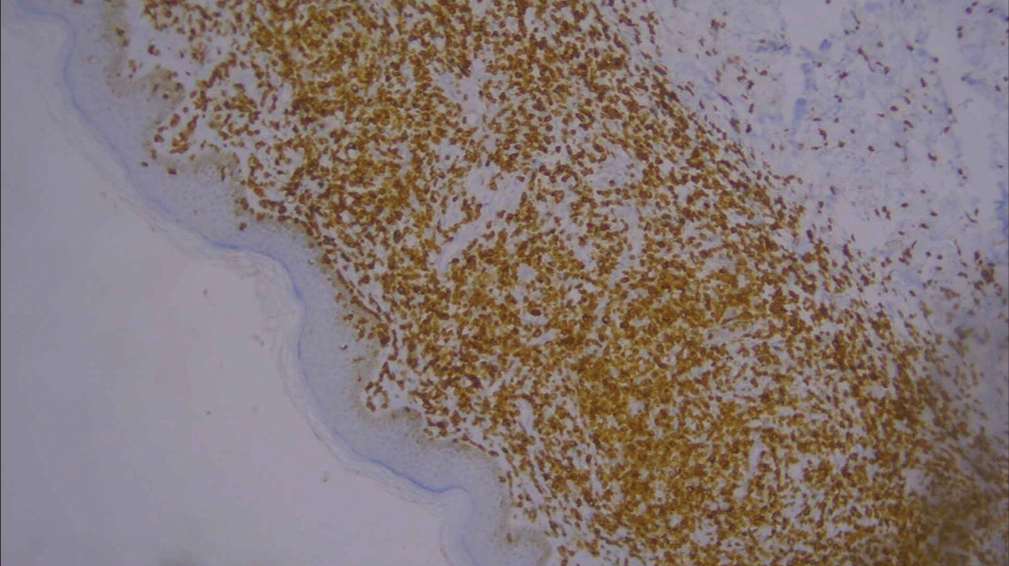
- Low-power image of immunohistochemistry showing CD3 positivity (10×).
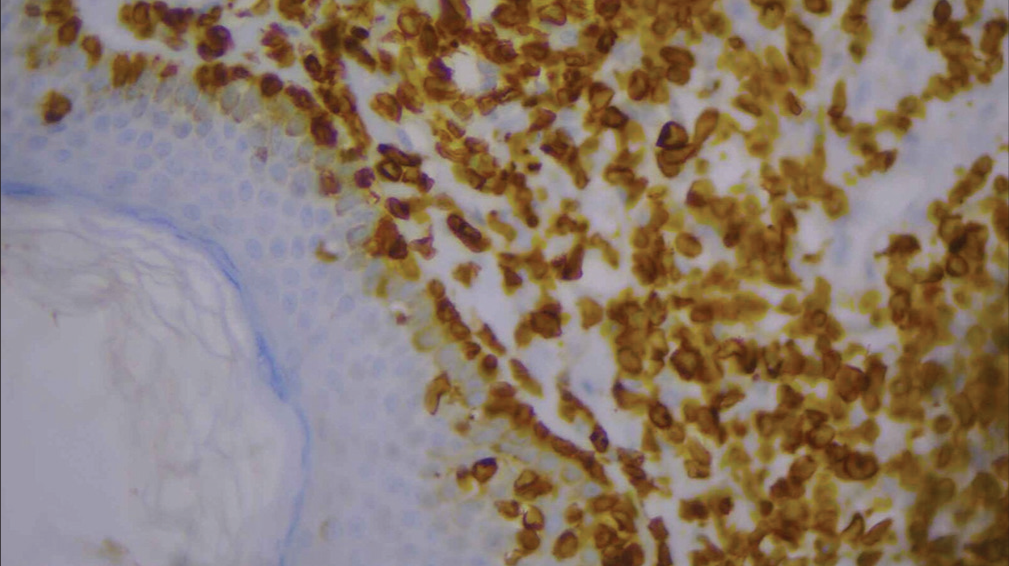
- High-power image of immunohistochemistry showing CD3 positivity (40×).
- High-power image of immunohistochemistry showing CD5 positivity (40×).
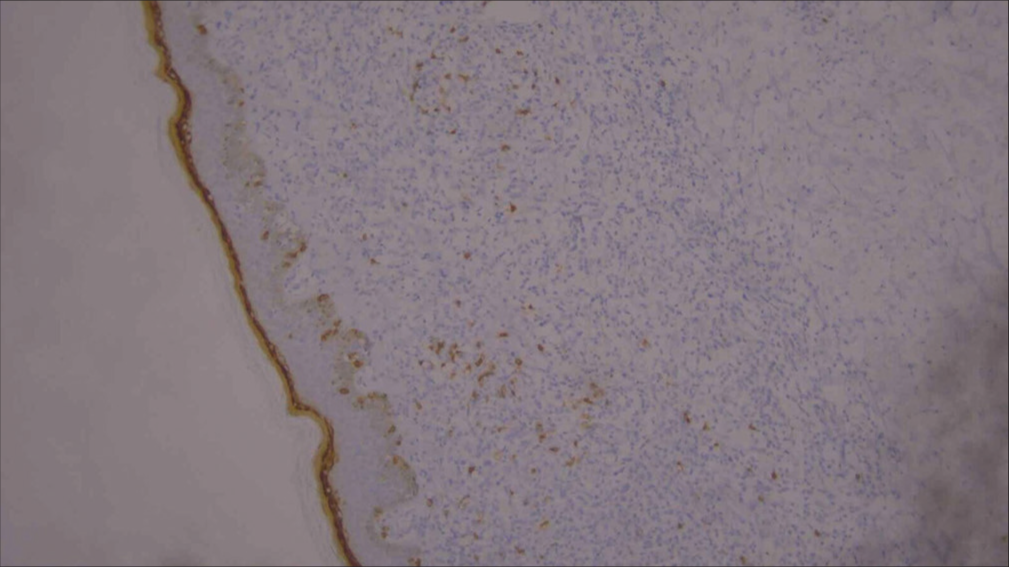
- Low-power image of immunohistochemistry showing CD8 positivity (10×).
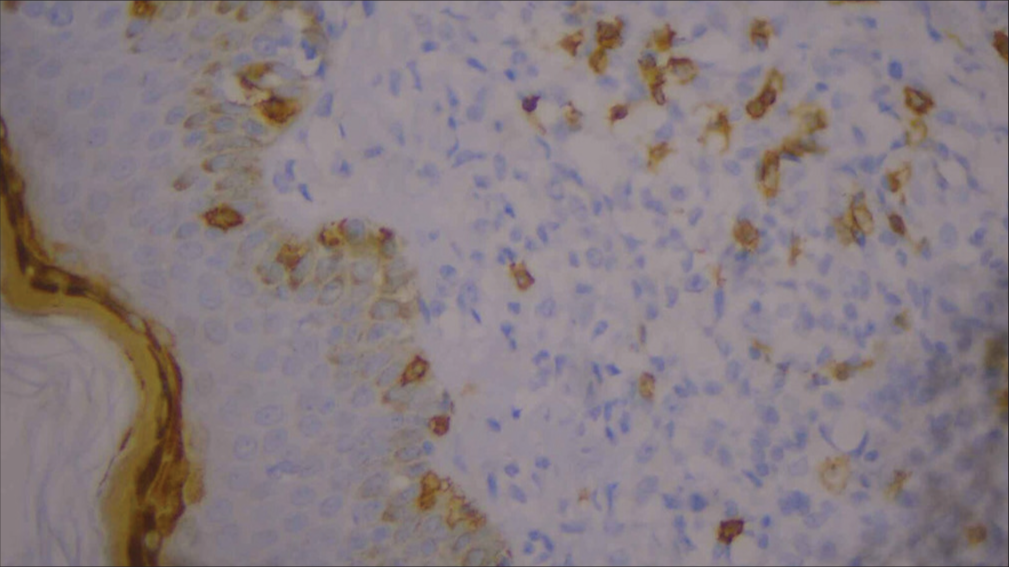
- High-power image of immunohistochemistry showing CD8 positivity (40×).
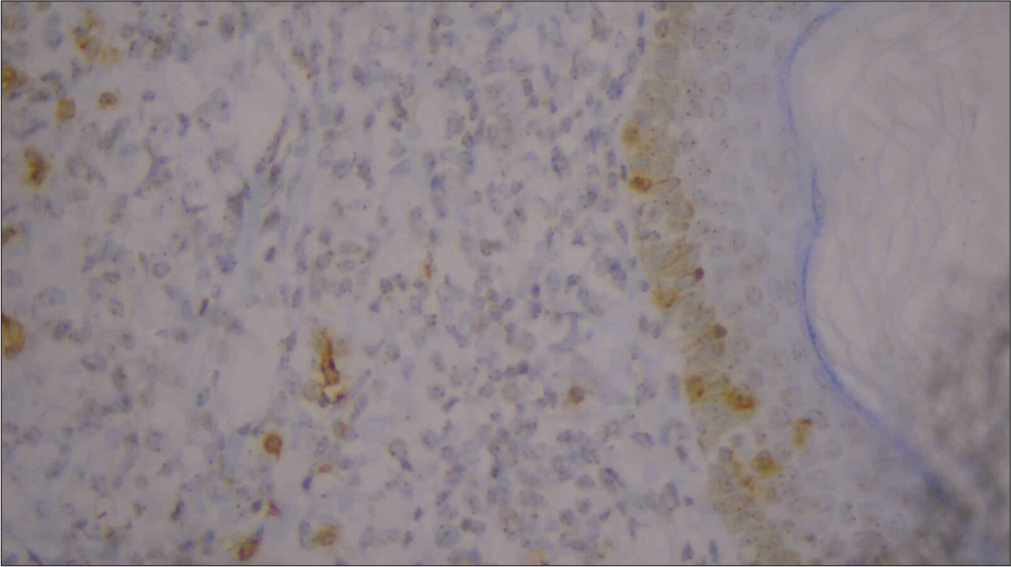
- High-power image of immunohistochemistry showing negative CD7 (40×).
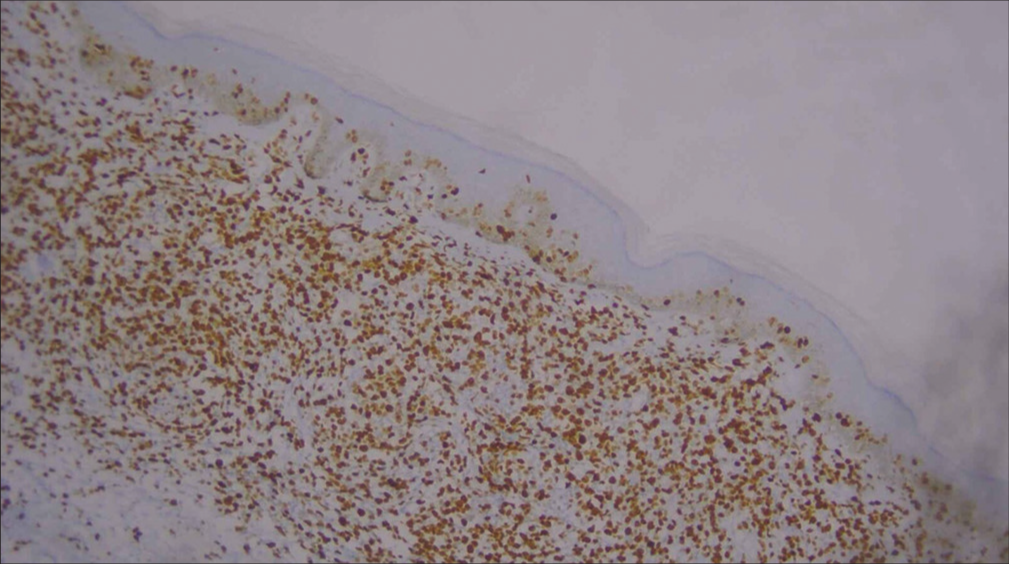
- Low-power image of Ki-67 immunohistochemistry (10×).
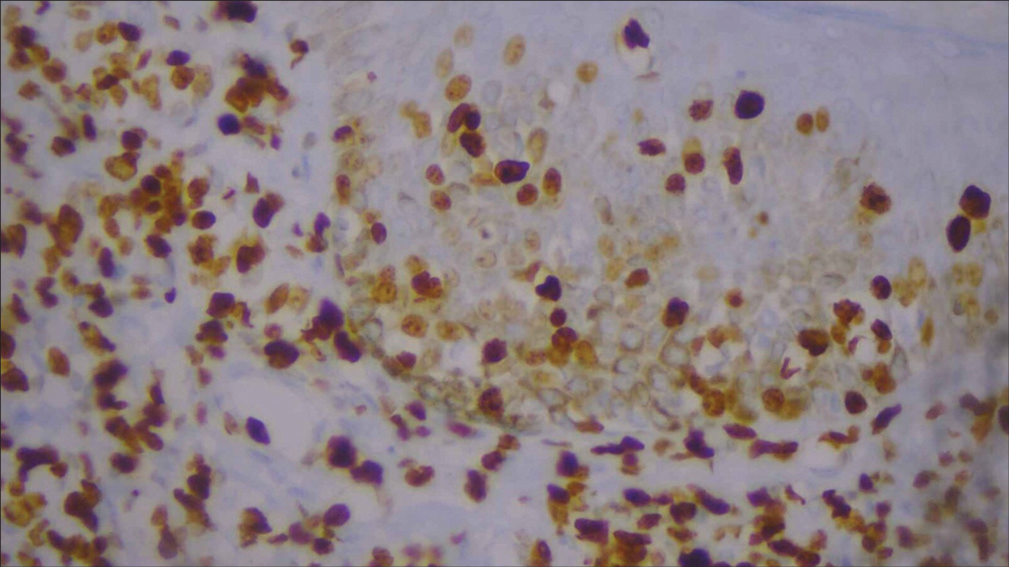
- High-power image of Ki-67 immunohistochemistry (40×).
The history, examination findings, histopathology findings, and IHC reports were confirmed on the diagnosis of early stage of MF.
He was treated with weekly blood transfusions, erythropoietin injections, oral pyridoxine, and oral folic acid for myelodysplastic neoplasm, to which he had symptomatic relief in the initial months of treatment.
MF was managed topical emollients, anti-histamines, and psoralen ultraviolet A (PUVA) therapy using oral trioxsalen ingestion, 2 h before phototherapy and ultraviolet A (UVA) phototherapy in the dose of 13–14 J/cm2 per session, done thrice weekly for a cycle of 28 days.
Follow-up was done monthly for one year and the skin lesions have improved significantly after five cycles of phototherapy, but the general health condition of the patient has deteriorated by worsening of fatigue and reduction in blood counts despite the supportive management for myelodysplastic neoplasm.
DISCUSSION
The most prevalent form of CTCL is MF, a low-grade lymphoproliferative disorder.[4] Approximately 40% of all cutaneous lymphomas are of the most frequent type, MF, which often manifests in mid-to-late adulthood (median age at diagnosis: 55–60 years) with a 2:1 male predominance.[4]
The WHO classifies MF as being characterized by its classic form, which includes patches, plaques, or variants.[5]
Neoplastic T-cells usually localize on the skin and cause erythroderma, plaques, patches, or tumors.[4]
The clinical variants of MF are folliculotropic/pilotropic, poikiloderma, hypopigmented, capillaritis-like, verrucous/hyperkeratotic, psoriasiform, ichthyosiform, and bullous.[6]
Due to the extremely varied presentations of MF and the non-specific characteristics of histological findings at times, the diagnosis can be challenging.[4]
When a patient is diagnosed, peripheral blood samples should be obtained for routine hematology, biochemistry, serum lactate dehydrogenase (LDH), Sézary cells, lymphocyte subsets, CD4:CD8 ratio, human T-lymphotropic virus type 1 (HTLV-1) serology, and, ideally, peripheral blood mononuclear cell T-cell receptor gene analysis.[6]
Depending on the clinical stage, MF has different histological characteristics.[6]
The papillary dermis has a moderate lymphocytic infiltration, which is one of the initial pathological characteristics of MF. Prominent epidermotropism, which is defined by the selective colonization of the epidermis by atypical T-cells, frequently along the basal layer, or by clusters of atypical lymphocytes in the epidermis known as Pautrier microabscesses, develops as the disease worsens and thicker plaques form.[6]
In addition to having a highly irregular nuclear outline, heavy nuclear staining, and a distinctive halo appearance, malignant T-cells in the epidermis are frequently cerebriform.[6]
Accurate diagnosis has been made possible by molecular biology. Given that MF can mimic a wide range of skin conditions, clinical experience is crucial.[4]
Topical steroids, carmustine, and topical chemotherapy with mechlorethamine (nitrogen mustard) are the available treatment options for MF.
Both UVB, PUVA and UVA-1 phototherapy, immunotherapy using interleukin-12, interferon (IFN)-a, and IFN-g, as well as radiotherapy including whole skin electron beam therapy and retinoids are also used in management of MF.[6]
Myelodysplastic syndromes, now known as myelodysplastic neoplasm, according to WHO HAEM5, are a group of clonal disorders originating from HSCs and typically exhibiting various degrees of cytopenia, dysplasia in one or more myeloid cell lineages, and ineffective hemopoiesis.[7,8]
The subclassification is based on percent of bone marrow and peripheral blood blasts, type/degree and number of dysplastic cell lineages, presence/absence of ring sideroblasts, and presence of specific chromosomal abnormalities.
The median age at the diagnosis of MDS is 71 years.[7] 10– 15% of MDS patients may progress to AML.[7]
The severity of the disease determines the treatment options for MDS patients.
Typically, low-risk MDS patients are recommended to receive supportive care that includes growth factors, transfusions, and antibiotic medication.[7]
In high-risk MDS patients, chemotherapy regimens include cytarabine, idarubicin, daunorubicin, immunomodulatory drugs (lenalidomide), hypomethylating treatment (HMT; decitabine and 5-azacytidine), and cytarabine are frequently utilized to delay development of AML.[7]
Ultimately, HSC transplantation and high-dose chemotherapy are used to establish long-term remission.[7]
There have been prior reports of lymphoid and myeloid cancers coexisting in the same patient.[9]
Certain occurrences happen without the use of chemotherapy, despite the fact that this is often linked to the leukemogenic effect of the myeloma treatment.[9]
In the past, patients who recovered from common lymphoid neoplasms were more susceptible to therapy-related myelodysplastic syndrome/AML (tMDS/AML).[10]
Patients with MF have a higher relative risk (1.73; range, 1.32–2.4) of developing secondary cancers compared to healthy individuals, and there have been reports of CTCL cooccurring with other hematoproliferative illnesses.[11]
Moritz et al., have reported a case of 65-year-old man with erythroderma and initially diagnosed with MF on histopathology, who responded to systemic PUVA treatment, and got relapse with worsening of symptoms with features consistent with chronic myelomonocytic leukemia, with clinical findings of splenomegaly and palpable lymphadenopathies and laboratory findings of leukocytosis, monocytosis, anemia, thrombocytopenia, and elevated LDH.[11]
In the patient described by Rostoker et al., MDS preceded the development of Sezary syndrome by 18 months.[9]
In this case, the patient initially had myelodysplastic neoplasm, and 9 months later, he developed MF and after multiple cycles of phototherapy (PUVA), he experienced worsening of MDS possibly due to the phototherapy or due to the natural course of the disease.
The coexistence of MF and myelodysplastic neoplasm could be due to various mechanisms such as a mutation in a common progenitor cell of the lymphoid and myeloid lineages, which results in stem cells that are unstable and run the risk of becoming cancerous myeloid and/or lymphoid clones, genetic processes that independently predispose cells of the B and T lineages to develop into lymphomas, treatment of the first neoplasm which provides an enhanced environment for the development of the second neoplasm, or could be the result of two unrelated tumors occurring by coincidence.[12,13]
In addition, it is hypothesized that these disorders may coexist due to a shared etiology such as solvents, chemicals, virus such as HTLV1 and Ebstein Barr virus, and carcinogen exposure, which influence B- and T-cell progenitors, trigger oncogenes, or deactivate tumor suppressor genes and produce dysplastic processes that impact lymphoid cell lines, which, in turn, cause immunologic disorders.[13,14]
Even though the true incidence of MF and pseudo-MF in small B-cell malignancies is not known, the occurrence has been postulated due to the neoplastic T- and B-cells of true MF and B-chronic lymphocytic leukemia derived from a common lymphocytic ancestor.[15]
CONCLUSION
This case contributes to the limited literature on the coexistence of MDS and MF. While therapy-related myelodysplastic neoplasm is known, this case emphasizes de novo occurrence, prompting further research into shared pathophysiological mechanisms.
The challenge lies in understanding the intricate interplay between these hematologic and dermatologic disorders for optimal therapeutic strategies. This report serves as a valuable addition to the medical community’s knowledge, urging continued exploration of such rare coexisting malignancies to enhance diagnostic precision and treatment efficacy.
Ethical approval
The Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Mycosis fungoides: Tumour d'emblee. Indian Dermatol Online J. 2012;3:122-4.
- [CrossRef] [PubMed] [Google Scholar]
- Baron Jean-Louis Alibert (1768-1837) and the first description of mycosis fungoides. J BUON. 2014;19:585-8.
- [Google Scholar]
- Myelodysplastic syndrome In: StatPearls. Treasure Island, FL: StatPearls Publishing; 2023.
- [Google Scholar]
- Mycosis fungoides: A dermatological masquerader. Br J Dermatol. 2007;156:1-10.
- [CrossRef] [PubMed] [Google Scholar]
- Cutaneous lymphoma In: Kang S, Amagai M, Bruckner AL, Enk AH, Margolis DJ, eds. Fitzpatrick's dermatology (9th ed). New York: McGraw-Hill; 2019. p. :2072.
- [Google Scholar]
- Cutaneous lymphomas: Mycosis fungoides In: Griffiths C, Barker J, Bleiker T, Chalmers R, Creamer D, eds. Rook's textbook of dermatology (9th ed). Oxford: Wiley-Blackwell; 2016. p. :3856.
- [Google Scholar]
- Pathogenesis of myelodysplastic syndromes: An overview of molecular and non-molecular aspects of the disease. Blood Res. 2014;49:216-27.
- [CrossRef] [PubMed] [Google Scholar]
- The 5th edition of the World Health Organization classification of haematolymphoid tumours: Lymphoid neoplasms. Leukemia. 2023;37:1944-51.
- [CrossRef] [PubMed] [Google Scholar]
- Coexistence of Sezary's syndrome and dysmyelopoiesis with an excess of myeloblasts. J Am Acad Dermatol. 1986;15:1296-8.
- [CrossRef] [PubMed] [Google Scholar]
- Trends in risk for therapy-related myelodysplastic syndrome/acute myeloid leukaemia after initial chemo/immunotherapy for common and rare lymphoid neoplasms, 2000-2018. EClinicalMedicine. 2023;61:102060.
- [CrossRef] [PubMed] [Google Scholar]
- Exacerbation of mycosis fungoides leading to the diagnosis of chronic myelomonocytic leukaemia. JAAD Case Rep. 2018;4:270-3.
- [CrossRef] [PubMed] [Google Scholar]
- Cutaneous T-cell lymphoma and myelodysplastic syndrome. JAAD Case Rep. 1994;31:1065-7.
- [CrossRef] [PubMed] [Google Scholar]
- Composite mycosis fungoides and B-cell chronic lymphocytic leukemia. Ann Diagn Pathol. 2002;6:172-82.
- [CrossRef] [PubMed] [Google Scholar]
- Stage II mycosis fungoides associated with myelodysplastic syndrome-case report. RoJCED. 2016;3:173-7.
- [Google Scholar]
- Folliculotropic T-cell infiltrates associated with B-cell chronic lymphocytic leukaemia or MALT lymphoma may reveal either true mycosis fungoides or pseudolymphomatous reaction: Seven cases and review of the literature. J Eur Acad Dermatol Venereol. 2015;29:77-85.
- [CrossRef] [PubMed] [Google Scholar]




