
Translate this page into:
Hypereosinophilic syndrome – Two cases with varied manifestations
*Corresponding author: Dr. Mary Vineetha, Department of Dermatology, Government Medical College, Kottayam, Kerala, India. drmaryvineetha@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Maniyan KM, Vineetha M, Celine MI, Sadeep MS, Kurien G, Anitha A. Hypereosinophilic syndrome – Two cases with varied manifestations. J Skin Sex Transm Dis 2019;1:41-4.
Sir,
Hypereosinophilic syndrome (HES) is a myeloproliferative disorder (MPD) characterized by persistent eosinophilia and damage to multiple organs.[1] First described by Hardy and Anderson, in 1968, HES is characterized by sustained overproduction of eosinophils. Mucocutaneous lesions can be the presenting feature in 25–50% of cases. Before making a diagnosis of HES, other causes of secondary eosinophilia need to be ruled out through appropriate investigations. Here, we report two cases of HES – coexistence of HES with bullous pemphigoid and HES presenting as vasculitis.
Case 1: A 55-year-old female patient presented with pruritic papules, vesicles, and tense bullae of 1-year duration. Lesions were distributed over erythematous as well as on normal-looking skin of extensor aspects of forearms, legs, trunk, and face [Figure 1]. They healed leaving post-inflammatory hypo-and hyper-pigmentation.
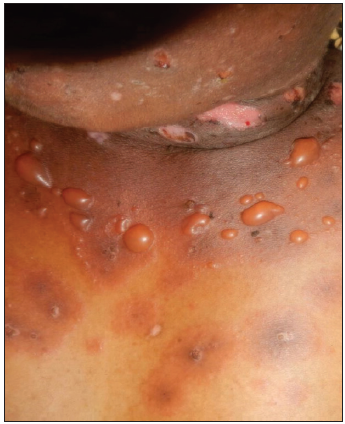
- Vesicles and tense bullae of bullous pemphigoid on an erythematous base on trunk.
The patient had non-tender submandibular and inguinal lymphadenopathy. Nikolsky’s sign and bulla spread sign were negative. Systemic examination was within normal limits.
Investigations revealed total count of 7800/mm3, low hemoglobin level (9 g%), elevated erythrocyte sedimentation rate (114 mm), and eosinophilia (57% in peripheral smear with an absolute eosinophil count [AEC] of 2700/dl). Peripheral smear analysis did not show any atypical cells. Tzanck smear showed plenty of neutrophils and eosinophils. Skin biopsy and direct immunofluorescence study were consistent with bullous pemphigoid [Figure 2]. No abnormality was detected in chest X-ray.
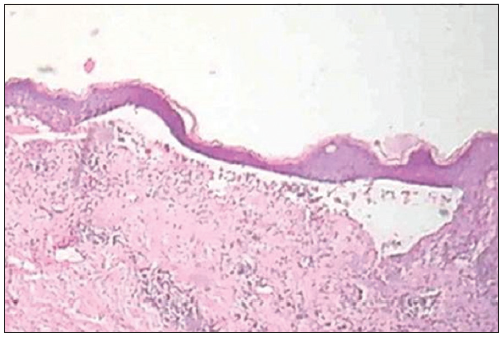
- Skin biopsy showing subepidermal blister (H and E, ×100).
The patient responded to oral prednisolone started at a dose of 1 mg/kg body weight. However, even after 1 month of steroid therapy, her AEC remained high at 3200/dl.
A thorough evaluation was carried out to detect the cause of persistent eosinophilia. Stool microscopy was within normal limits. Serum electrophoresis revealed polyclonal gammopathy with highly elevated gamma globulin fraction. Bence Jones proteins were absent in urine and skull X-ray was within normal limits.
Excision biopsy of inguinal lymph node revealed reactive hyperplasia with plasmacytosis. Bone marrow aspirate showed trilineage leukocytosis with increased eosinophil precursors and mild plasmacytosis. Reactive lymphoid hyperplasia and plasmacytosis without any evidence of lymphoma were the findings in immunohistochemistry study of lymph node biopsy specimen. Contrast-enhanced computerized tomogram of chest and bronchial lavage examination ruled out pulmonary eosinophilia and lymphoma.
A final diagnosis of idiopathic HES coexisting with bullous pemphigoid was made. Echocardiogram recorded normal study. The patient was continued on oral prednisolone that was slowly tapered 5 mg every month. Dapsone 100 mg/day, diethylcarbamazine 100 mg 3 times daily for 21 days, and two tablets of albendazole 400 mg 1 week apart were added to systemic steroids. The patient was kept under regular follow-up and was evaluated every 6 months with peripheral smear and bone marrow analysis and ultrasound examination of abdomen.
Dapsone has to be withdrawn after 5 months due to persistent anemia which was found to be hemolytic in nature. Hydroxyurea 500 mg tablet once a day was initiated as advised by physician for persistent eosinophilia and also for controlling the new-onset bullous pemphigoid lesions that appeared with tapering of steroids. 1 month after starting hydroxyurea, her AEC level came down to 290/dl. She is currently receiving prednisolone 15 mg and hydroxyurea 250 mg (tapered after 3 months).
Our second case was a 44-year-old female who presented with reddish lesions on legs of 3 weeks duration. On examination, she had retiform purpura on both legs associated with gangrenous changes of fingers [Figures 3 and 4]. She was evaluated with a provisional diagnosis of polyarteritis nodosa. Her investigations revealed leukocytosis and eosinophilia of 40% (AEC 6048/ul) without any atypical cells or blast forms in peripheral smear analysis.
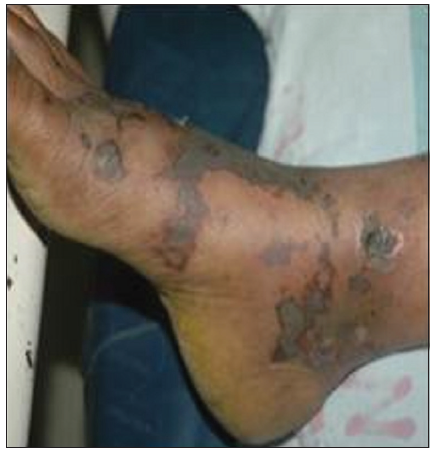
- Retiform purpura on foot.
Investigation panel for retiform purpura including cryoglobulins, antinuclear antibody, antiphospholipid antibodies, antineutrophil cytoplasmic antibodies (ANCA), and markers for viral hepatitis was negative. Causes of eosinophilia were ruled out by detailed evaluation.
While in hospital, she developed right-sided wrist drop. Nerve conduction study revealed axonopathy of radial nerve. She was prescribed intravenous dexamethasone at a dose of 1 mg/kg prednisolone equivalent which was later changed to oral prednisolone which was tapered at a dose of 5 mg once a month. Skin lesions subsided and nerve function improved. Histopathology revealed eosinophilic vasculitis with leukocytoclasia and organizing thrombus [Figure 5]. Persistent eosinophilia (2400/dl) even after 2 months favored the possibility of HES presenting as vasculitis. Further, evaluation showed involvement of no other systems. The patient is currently under regular follow-up.
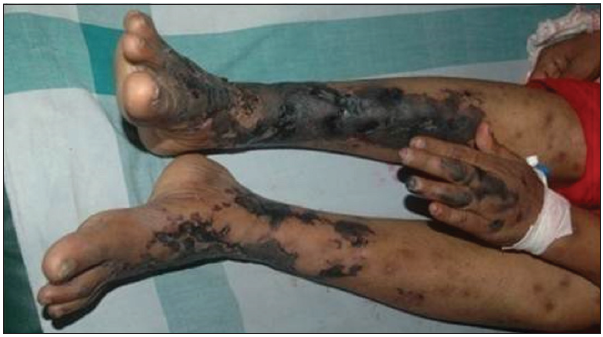
- Retiform purpura on legs and gangrene of index finger.
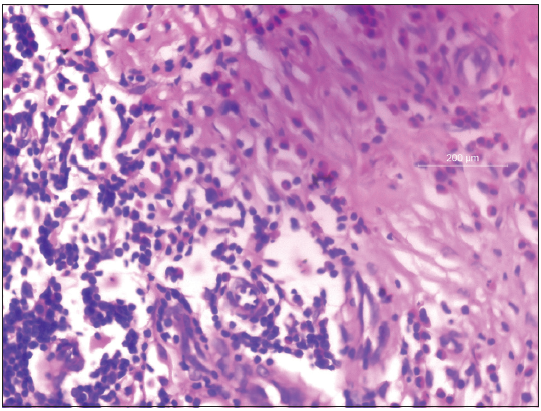
- Skin biopsy from retiform purpura showing eosinophilic vasculitis (H and E, ×400).
As per the revised diagnosed criteria for HES, peripheral blood eosinophilia >1500 mm3 on at least two occasions or evidence of prominent tissue eosinophilia favors a diagnosis of the same. Other causes of eosinophilia such as parasitic infestations, viral infections, allergic diseases, hypoadrenalism, neoplasms, drugs, or chemicals should be ruled out.[1]
The revised criteria are a modification of the original Chusid et al. criteria which required persistent eosinophilia of 6 months to diagnose HES.[2]
HES is classified into myeloproliferative, lymphocytic, undefined, familial, overlap, and associated types.
Myeloproliferative HES is often seen in males, may or may not be associated with positive FIP1L1-PDGFRA mutation and commonly accompanied by cardiac involvement. Increased mast cells and elevated tryptase levels are the other features. A diagnosis of chronic eosinophilic leukemia is made in the setting of FIP1L1-PDGFRA mutation.
In contrast to myeloid HES, lymphocytic HES shows no sex predilection. These patients often show predominant cutaneous manifestations including pruritus, eczema, erythroderma, urticaria, and angioedema. In general, patients with lymphocytic HES do not manifest cardiac disease.[3]
Mucocutaneous manifestations described in HES are pruritus, angioedema, urticaria, purpuric macules, papules, plaques, nodules, mucosal ulcers, gangrene, livedo reticularis, Raynaud’s phenomenon, vesicles, eczematous lesions, and vasculitis.[4] Apart from the heart (40% of cases), other systems affected are nervous, pulmonary, and gastrointestinal systems. Endomyocardial fibrosis is the end result of cardiac involvement in HES.[5]
Our first patient was thoroughly evaluated for other causes of eosinophilia before we arrived at a diagnosis of coexistence of HES and bullous pemphigoid which is already described in literature.[6]
The second patient had retiform purpura, gangrene, and mononeuritis multiplex that mimicked polyarteritis nodosa; but, histopathology favored HES. Jang et al. reported two cases of HES presenting with eosinophilic vasculitis and gangrene.[7] Our patient improved well with systemic steroids. Negative p-ANCA and allergic manifestations were against the possibility of Churg-Strauss syndrome. The therapy for symptomatic HES aims to debulk the blood and tissue eosinophil burden. Corticosteroids remain the cornerstone of treatment. Other immunosuppressants tried are hydroxyurea, interferon alfa, cladribine, cyclosporine, imatinib mesylate, mepolizumab, and alemtuzumab.[8]
HES should be a differential diagnosis in all cases of persistent eosinophilia. A full workup is mandatory before making a diagnosis of HES. Failure to detect the condition early may lead to irreversible cardiac damage. In view of its potential to involve other systems, a multidisciplinary approach is essential in managing HES. Gene analysis for FIP1L1-PDGFRA fusion protein should be done in therapy resistant and suspicious cases to rule out chronic eosinophilic leukemia. Patients should be kept under regular follow-up for early detection of other MPD or lymphoma.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patients have given their consent for their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Refining the definition of hypereosinophilic syndrome. J Allergy Clin Immunol. 2010;126:45-9.
- [CrossRef] [PubMed] [Google Scholar]
- The hypereosinophilic syndrome: Analysis of fourteen cases with review of the literature. Medicine (Baltimore). 1975;54:1-27.
- [CrossRef] [Google Scholar]
- The hypereosinophilic syndrome current concepts and treatments. Br J Haematol. 2009;145:271-85.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical overview of cutaneous features in hypereosinophilic syndrome. Curr Allergy Asthma Rep. 2012;12:85-98.
- [CrossRef] [PubMed] [Google Scholar]
- Cardiovascular features of 11 patients with eosinophilic endomyocardial disease. Q J Med. 1983;52:23-39.
- [Google Scholar]
- Bullous pemphigoid associated with hypereosinophilic syndrome: Simultaneous response to imatinib. J Am Acad Dermatol. 2007;56(Suppl 5):S68-72.
- [CrossRef] [Google Scholar]
- Hypereosinophilic syndrome presenting as cutaneous necrotizing eosinophilic vasculitis and Raynaud’s phenomenon complicated by digital gangrene. Br J Dermatol. 2000;143:641-4.
- [CrossRef] [PubMed] [Google Scholar]
- Approaches to the treatment of hypereosinophilic syndromes: A workshop summary report. J Allergy Clin Immunol. 2006;117:1292-302.
- [CrossRef] [PubMed] [Google Scholar]




