
Translate this page into:
Pathogenesis of atopic dermatitis: Current concepts
*Corresponding author: Jayamini Seneviratne, Department of Dermatology, Lady Ridgeway Hospital for Children, Colombo, Sri Lanka. dermalrh@gmail.com
-
Received: ,
Accepted: ,
Read UPDATED-ARTICLE associated with this - 10.25259/JSSTD_8_2021_ER
How to cite this article: Seneviratne J. Pathogenesis of atopic dermatitis: Current concepts. J Skin Sex Transm Dis 2021;3:113-7.
Abstract
Atopic dermatitis is the most common, yet more troublesome, inflammatory skin disease affecting mankind and its prevalence is increasing globally. In established disease, inflammation and pruritus dominate the clinical picture. Thanks to a group pioneering of dermatological scientists, we are now beginning to understand how inflammation is initiated by a primary defect in the epidermal skin barrier.
Keywords
Atopic dermatitis
Pathogenesis
Epidermis
Barrier defect
Inflammation

INTRODUCTION
Atopic dermatitis (AD) is the most common, yet more troublesome, inflammatory skin disease affecting mankind and its prevalence is increasing globally. In established disease, inflammation and pruritus dominate the clinical picture. Other associated features include xerosis, a propensity to develop specific skin infections, and an association with mucosal allergy. Thanks to modern research, the underlying basis of this complex disease is beginning to unveil. As inflammation is primarily a defense mechanism and mediated by both innate and adaptive immune systems, an immunological basis for the illness is now well accepted. Thanks to a group pioneering of dermatological scientists, we are now beginning to understand how inflammation is initiated by a primary defect in the epidermal skin barrier.
ROLE OF THE EPIDERMIS
Skin is primarily an organ of protection. To protect the milieu interior from a wide variety of harmful agents, the epidermis is both structurally and functionally well geared, hence the term skin barrier. It also prevents evaporation of water (transepidermal water loss -TEWL). Therefore, the latter reflects the integrity of the epidermal barrier. This function is predominantly carried out by the stratum corneum (SC) and rest of the epidermis, contributes by generating the SC.[1-5]
ROLE OF THE SC
The brick and mortar arrangement of SC provides a comprehensive biological and immunological barrier. Corneocytes (bricks) are protein-rich cells with storage facilities and without any synthetic functions. The mortar consists predominantly of a variety of lipids (ceramides, free fatty acids, and cholesterol). As keratinocytes move toward the surface to become corneocytes, they secrete the contents of the lamellar bodies which make up the mortar. The physical arrangement of the cornified envelope, cornified lipid envelope, and lamellar membranes provides a physical frame (scaffold) that regulates and strengthens the barrier. It also provides a physical base for various enzymes to act and facilitates the movement of fat-soluble molecules. Evaporation of water and ingress of allergen and antigens are prevented by the epidermal barrier. Thus, the skin barrier provides a comprehensive barricade function in protecting the dermis from harmful exterior milieu.[2,4-6]
WHAT CAUSES THE BARRIER DEFECTS IN AD
Both inherited defects and acquired insults are now established as primary events in barrier dysfunction. The fact that AD may start as early as 3 months of age is indicative of a failure of compensatory mechanisms involving barrier restoration.[7,8] The human fetus develops in a protective aqueous environment in utero. However, after birth, the baby is wrapped in all sorts of clothes, washed with alkaline soaps and shampoo, and covered in synthetic scented baby creams. All of the above products are proven to be harmful to the tender skin barrier of the baby.[2] This partly explains its onset at 3 months of age and its high prevalence in affluent families of the society.[7,9] It is well known that leave-on bland emollients delay the onset of the disease and also reduce the severity.[10].SC hydration plays a key role in barrier homeostasis through two mechanisms, exogenous and endogenous. Endogenous hydration is by prevention of evaporation of water and by retention of natural moisturizing factors (NMF) (see below).[10] Exogenous hydration is by bathing (immersion in water - a shortened version of in utero environment is created) and by use of occlusive emollients, which aid by trapping water in SC.[11]
INHERITED DEFECTS OF THE SKIN BARRIER IN AD
Filaggrin mutation was the first defect discovered.[12,13] Filaggrin derived from profilaggrin plays a key role in barrier homeostasis. First, filaggrin metabolites provide an essential barrier function by providing molecules for the NMF. These include lactic acid, amino acids, urea, inorganic salts, sugars, pyrrolidone carboxylic acid, and urocanic acid. NMF plays a key role in SC hydration and therefore homeostasis, in maintaining plasticity of the skin and in the process of enzymatic corneodesmolysis.[2-5] Therefore, deficiencies of these molecules lead to xerosis seen in AD. Second, filaggrin metabolites provide the additional vital function of supplying pyrrolidone carboxylic acid and urocanic acid that constitute the acid mantle.[4] The best SC function occurs at a pH of 5.5.[14] Thirdly filaggrin provides vital support to the keratinocyte cytoskeleton. Severe deficiency of filaggrin results in retraction of keratinocyte cytoskeleton, which, in turn, reduces the delivery of lamellar body lipids to the mortar.[4,15] Ichthyosis vulgaris is a well-known association of AD. Since it is an autosomal dominant condition, majority inherit ichthyosis vulgaris in a heterozygous manner. Those who inherit it in a homozygous manner develop AD early and develop a protracted illness which may persist into adulthood.[2] A spectrum of other inherited defects of cornified envelope, cornified lipid envelope, and lamellar membranes is known to result in AD. An example of this is Netherton syndrome.[2]
HOW AN EPIDERMAL BARRIER DEFECT LEADS TO DERMAL INFLAMMATION IN AD?
As the name implies, features of inflammation, are commonly observed, especially in acute flares of AD and the immunological basis for inflammation has gained acceptance worldwide. The proven efficacy of a wide variety of immune-suppressive agents in AD (used both topically and systemically) supports the immunological basis for the disease. Further dupilumab, an interleukin (IL)-4 blocker is now licensed for use in moderate to severe AD.[16,17] Wide arrays of similar biologics targeting IL-13, IL-31, and thymic stromal lymphopoietin (TSLP) are becoming popular therapies for AD among dermatologists.[18] TSLP, a cytokine secreted by keratinocytes on scratching, is now known to propagate AD by inducing myeloid dendritic cells, T helper Type 2 (Th2) responses, and mast cell degranulation leading to cytokine secretion.[3,19]As shown in Figure 1, TSLP also plays a crucial role in itch-scratch cycle.[20] The new biologics target mainly inflammation. Hence, with the cessation of treatment, the disease re-emerges. It is also observed that emollients are useful ancillary therapy along with anti-inflammatory therapies whether they are topical or systemic agents or biologics. These points highlight the complexities in the pathogenesis of AD [Figure 1].
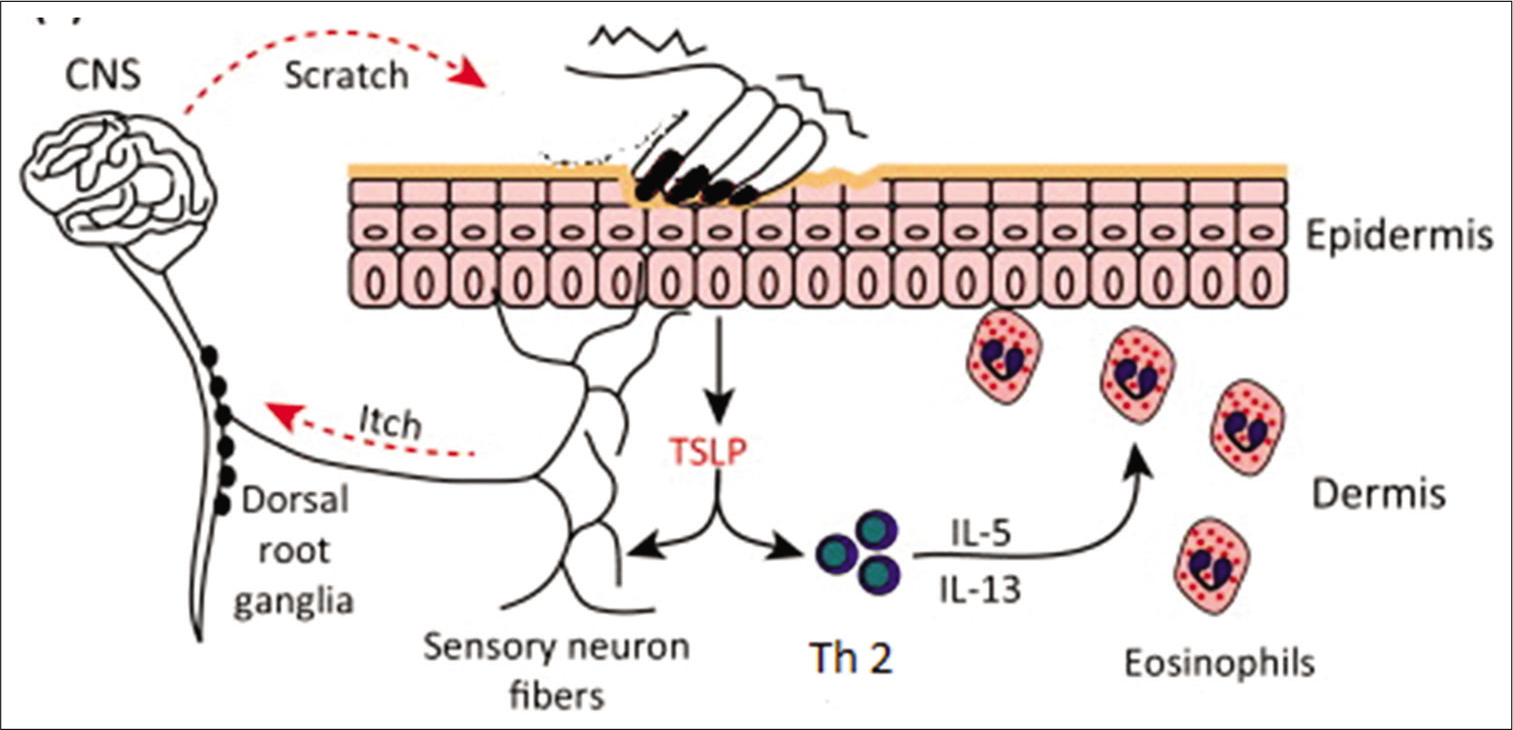
- Diagramatic representation of pathogenesis of atopic dermatitis.
- CNS: Central nervous system, Th 2: Type 2 helper T cell, IL: Interleukin.
The above has clearly lent support to the theory that inflammation in AD is directly linked to events taking place in the epidermis (sustained barrier dysfunction). Once the permeability barrier is perturbed in a sustained manner, a number of defense mechanisms are activated to protect the underlying dermis. On one side, pro-inflammatory cytokines are released while on the other side epidermal defense responses are generated. The latter is aimed at barrier restoration (barrier recovery).[4]
However, a sustained defect in the epidermal permeability barrier impairs the homeostatic response, allows the inflammation to spread and, recruits inflammatory cells consisting of lymphocytes, eosinophils, and mast cells.[2] However, neutrophils are usually absent.[21]
The defective barrier allows the entry of many noxious agents including environmental antigens, aeroallergens, and haptens as well as antigens derived from microorganisms, especially Staphylococcus aureus.[2,4] Some toxins of S. aureus act as superantigens that rapidly accelerate the establishment of inflammation.[4,8] Allergens originating from house dust mite in particular, promote protease secretions, further enhancing the secretion of inflammatory cytokines.[4,5] The persistence of this process switches the specific protective adaptive immune responses to a more allergy dominant Th2 and Th22 dominant immune profile.[4] A fair proportion of patients develop high IgE antibodies and IL-31 secreting lymphocytes. This inflammation results in further deterioration of the permeability barrier.[4] As a result, barrier function is further compromised and barrier recovery is delayed. This further worsens the dermal inflammation resulting in a vicious cycle. Once established the inflammation runs a prolonged course, characterized by remission and relapses. Acute flares may result from super colonization by S. aureus.[4] Prolonged inflammation, especially when widespread, is very detrimental to the health of the individual. This is especially so in children. The catabolism of inflammation contributes partly to the growth retardation seen in children afflicted with AD. Other factors that contribute to growth retardation in atopic children are increased TEWL (which is energy-dependent), reduced secretion of growth hormones due to nocturnal pruritus, poor appetite due to inflammation of the disease, and associated infections.[22-24]
PROPENSITY TO DEVELOP SKIN INFECTIONS
The skin microbiome consists of a number of microorganisms that cannot be detected by routine culture methods. In AD marked changes are observed in the microbiome.[2,4,5] This consists of dominant colonization by S. aureus and epidermidis. Many factors contribute to this. These include reduced expression of beta-defensins, relative deficiencies of anti-infectious Th1 cytokines, and elevated pH that occurs in active AD.[2,4,5] Similarly, improvement in barrier function leads to restoration of normal flora demonstrating a close relationship between permeability barrier and antimicrobial barrier. However, this is not on an equal basis. Dermatologists are familiar with eczema verrucatm and eczema molluscatum in which inflammation is hardly noticed. Certain infections result in acute flares (e.g., S. aureus) whereas some lead to life-threatening situations (eczema herpeticum).[2,4,25]
PRURITUS
A disease defining feature of AD is pruritus. It is one of the most troublesome aspects of the disease. Hence, pruritus can be used to assess the efficiency of treatment of AD. In other words, AD is a typical model of chronic pruritus. Pruritus which is worse at night leads to sleep disturbance, daytime somnolence, depression, anxiety, learning disability, and in the case of children growth retardation due to reduced secretion of growth hormones. Once established, pruritus runs in a vicious cycle, the so-called itch-scratch-itch cycle [Figure 2].[26]
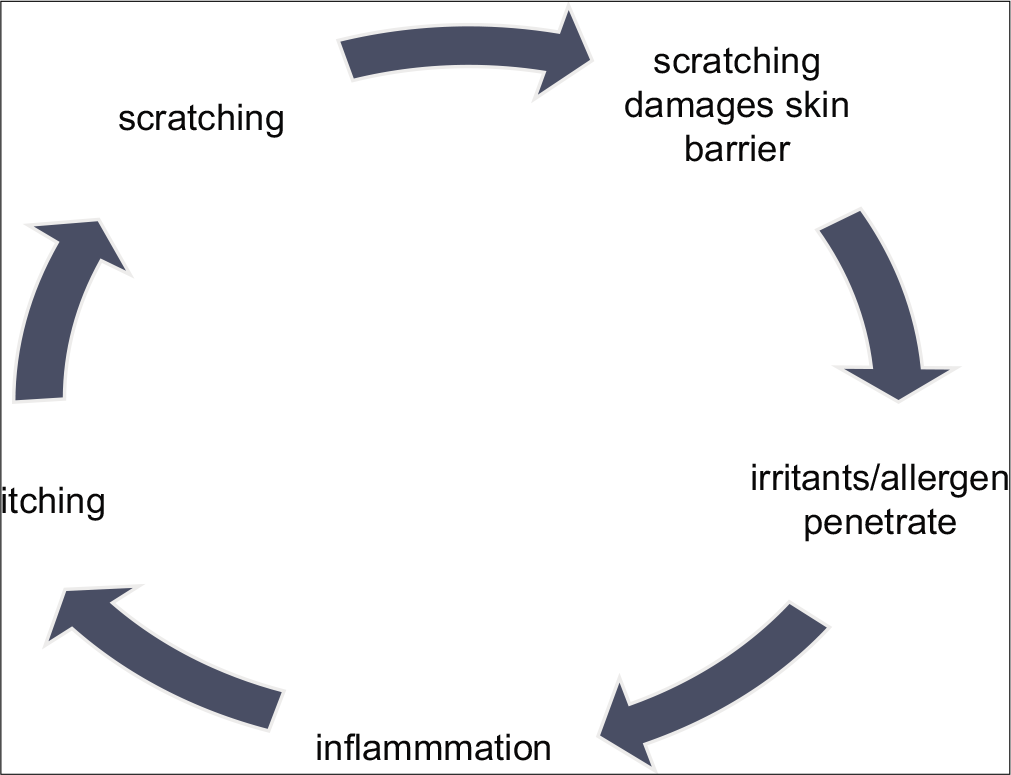
- Itch-scratch-itch cycle.
ROLE OF EPIDERMIS IN ATOPIC ITCH
It is well-known that itch originates from the itch-specific nerve fibers in the epidermis and dermo-epidermal junction. These nerve fibers (C fibers) reach keratinocytes and extend up to the stratum granulosum.[26]
EPIDERMAL BARRIER DYSFUNCTION AND ITCH
Intensity of pruritus in AD is associated with TEWL, which in return reflects the barrier integrity. Hence, severe barrier defect is associated with severe pruritus.[26] TEWL is known to increase epidermal pH, with resultant accumulation of SC serine protease and SC chymotryptic enzyme. These enzymes are known pruritogens, receptors for which are sensitized and up-regulated in chronic AD. The dysfunctional epidermal barrier promotes the ingress of environmental aeroallergens and irritants which are also pruritogens. There is also an association between barrier dysfunction and the development of food allergy. Exposure of skin and ingestion of food allergen lead to sensitization and production of specific IgE to these allergens. This was well shown in a recent study by Brough et al.[27] In this study, there was an exposure-response relationship between peanut protein in household dust and peanut specific skin prick test positivity. This also explains why AD history and severity are known risk factors for peanut allergy. Environmental irritants (soaps, perfumes, clothes, and metals) are known to directly activate transient receptor potential (TRP) channels on the keratinocytes.
Thermo-heat receptors TRP ankyrin 1 (TRPA1) expression is upregulated in AD skin lesions and its activation leads to the release of inflammatory cytokines and molecules that promote pruritus. TRPA1 is implicated as a transducer of non-histaminergic itch of the central nervous system and a mediator of neurogenic inflammation.[28] IL-31 plays a critical role in linking the neurons with inflammation induced by Th2 cells.[29] Expression of TRPA1 is markedly elevated in keratinocytes, sensory nerve endings, and mast cells of AD cells.[28] It is also implicated in acute flares of inflammation noted in itch-scratch cycles that are provoked by xerosis. This also explains the soothing effects seen when emollients are regularly used. In other words, TRPA is a major sensor of skin barrier perturbation.
The binding of these molecules to receptors on the surface of keratinocytes not only stimulates the latter but also leads to the transmission of sensation similar to axonal transport. This clearly identifies keratinocytes as the key player in AD.[30,31] Dermatologists are quite familiar with the troublesome itching seen in patients with chronic AD. Often this is intractable in lichenified areas. Apart from the initiating role, keratinocytes are responsible for conveying the itch sensation to cutaneous nerve fibers.
NEURONAL HYPERSENSITIVITY IN ATOPIC ITCH
As AD progresses, the itch becomes chronically entrenched as one of the most troublesome aspects of the disease. Chronic sufferers of AD report increased sensitivity to itch to previously non-itchy stimuli (allokinesis).[32] These can only be explained as due to the increased sensitivity of the peripheral nerve neural pathway. This happens either by hyper-innervation or by a decrease in itch threshold or both. Similar changes are also known to occur in the brain. Brain images in chronic AD sufferers have demonstrated increased activities in both the anterior cingulate areas of the prefrontal cortex.[33] Sudden intense itch noted in AD patients when irritants (like wool) come in contact can be easily explained by this theory. Furthermore, it can be demonstrated that the patients with AD have a higher density of cutaneous nerve fibers; moreover, the nerve fibers in AD patients are much larger in size.[34,35]
CONCLUSION
A better understanding of the intricate pathogenesis of atopic dermatitis may help to formulate effective treatment guidelines for this challenging disease.
Acknowledgment
Dr. M R F Shireen, Senior Registrar in Dermatology, Lady Ridgeway Hospital for children, Colombo.
Declaration of patient consent
Not required as there are no patients in this article.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Controlling the hydration of the skin through the application of occluding barrier creams. J R Soc Interface. 2013;10:20120788.
- [CrossRef] [PubMed] [Google Scholar]
- Mechanisms of abnormal lamellar body secretion and the dysfunctional skin barrier in patients with atopic dermatitis. J Allergy Clin Immunol. 2014;134:781-91.
- [CrossRef] [PubMed] [Google Scholar]
- Skin barrier abnormalities and immune dysfunction in atopic dermatitis. Int J Mol Sci. 2020;21:2867.
- [CrossRef] [PubMed] [Google Scholar]
- Skin barrier defects in atopic dermatitis. Curr Allergy Asthma Rep. 2014;14:433.
- [CrossRef] [PubMed] [Google Scholar]
- Skin barrier in atopic dermatitis: Beyond filaggrin. An Bras Dermatol. 2016;91:472-8.
- [CrossRef] [PubMed] [Google Scholar]
- Lipids and the permeability and antimicrobial barriers of the skin. J Lipids. 2018;2018:5954034.
- [CrossRef] [PubMed] [Google Scholar]
- Atopic dermatitis: Early treatment in children. Curr Treat Options Allergy. 2017;4:355-69.
- [CrossRef] [PubMed] [Google Scholar]
- Atopic dermatitis: A disease of altered skin barrier and immune dysregulation. Immunol Rev. 2011;242:233-46.
- [CrossRef] [PubMed] [Google Scholar]
- Do early-life exposures explain why more advantaged children get eczema? Findings from the U.K. millennium cohort study. Br J Dermatol. 2016;174:569-78.
- [CrossRef] [PubMed] [Google Scholar]
- Is endogenous glycerol a determinant of stratum corneum hydration in humans? J Invest Dermatol. 2005;125:288-93.
- [CrossRef] [PubMed] [Google Scholar]
- Use of emollients in atopic dermatitis. J Eur Acad Dermatol Venereol. 2015;29:854-7.
- [CrossRef] [PubMed] [Google Scholar]
- Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 2006;38:441-6.
- [CrossRef] [PubMed] [Google Scholar]
- Loss-of-function variations within the filaggrin gene predispose for atopic dermatitis with allergic sensitizations. J Allergy Clin Immunol. 2006;118:214-9.
- [CrossRef] [PubMed] [Google Scholar]
- The pH of the skin surface and its impact on the barrier function. Skin Pharmacol Physiol. 2006;19:296-302.
- [CrossRef] [PubMed] [Google Scholar]
- Primary role of barrier dysfunction in the pathogenesis of atopic dermatitis. Exp Dermatol. 2018;27:847-51.
- [CrossRef] [PubMed] [Google Scholar]
- Efficacy and safety of dupilumab in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical treatments: A randomised, placebo-controlled, dose-ranging phase 2b trial. Lancet. 2016;387:40-52.
- [CrossRef] [Google Scholar]
- 2019. Available from: https://www.drugs.com/newdrugs/fda-approves-dupixent-dupilumab-moderate-severe-atopicdermatitis-adolescents-4929.html [Last accessed on 2019 Sep 24]
- Biologics for allergic dermatologic diseases. Curr Allergy Asthma Rep. 2020;20:35.
- [CrossRef] [PubMed] [Google Scholar]
- The role of thymic stromal lymphopoietin in the immunopathogenesis of atopic dermatitis. Clin Exp Allergy. 2011;41:1515-20.
- [CrossRef] [PubMed] [Google Scholar]
- The spongiotic reaction pattern In: Weedon's Skin Pathology (3rd ed). Ch. 5. London: Churchill Livingstone; 2010. p. :94-122.
- [CrossRef] [Google Scholar]
- Pathophysiologic basis for growth failure in children with ichthyosis: An evaluation of cutaneous ultrastructure, epidermal permeability barrier function, and energy expenditure. J Pediatr. 2004;145:82-92.
- [CrossRef] [PubMed] [Google Scholar]
- Growth Retardation in Children with Atopic Dermatitis. 2016. Munich GRIN Verlag. Available from: https://www.grim.com/document/340946 [Last accessed on 2016 Sep 27]
- [Google Scholar]
- Systemic inflammation, growth factors, and linear growth in the setting of infection and malnutrition. Nutrition. 2017;33:248-53.
- [CrossRef] [PubMed] [Google Scholar]
- Atopic eczema In: Griffiths C, Barker J, Bleiker T, Chalmers R, Creamer D, eds. Rook's Textbook of Dermatology (9th ed). United Kingdom: Wiley Blackwell; 2016. p. :41.1-34.
- [Google Scholar]
- Pathophysiology of pruritus in atopic dermatitis: An overview. Exp Dermatol. 2002;11:12-24.
- [CrossRef] [PubMed] [Google Scholar]
- Atopic dermatitis increases the effect of exposure to peanut antigen in dust on peanut sensitization and likely peanut allergy. J Allergy Clin Immunol. 2015;135:164-70.
- [CrossRef] [PubMed] [Google Scholar]
- TRPA1-dependent pruritus in IL-13-induced chronic atopic dermatitis. J Immunol. 2013;191:5371-82.
- [CrossRef] [PubMed] [Google Scholar]
- IL-31: A new link between T cells and pruritus in atopic skin inflammation. J Allergy Clin Immunol. 2006;117:411-7.
- [CrossRef] [PubMed] [Google Scholar]
- Biomarkers for itch and disease severity in atopic dermatitis. Curr Probl Dermatol. 2011;41:136-48.
- [CrossRef] [PubMed] [Google Scholar]
- Peripheral and central mechanisms of itch. Neuron. 2018;98:482-94.
- [CrossRef] [PubMed] [Google Scholar]
- Exacerbating factors of itch in atopic dermatitis. Allergol Int. 2017;66:8-13.
- [CrossRef] [PubMed] [Google Scholar]
- Distinct patterns of brain activity evoked by histamine-induced itch reveal an association with itch intensity and disease severity in atopic dermatitis. Br J Dermatol. 2009;161:1072-80.
- [CrossRef] [PubMed] [Google Scholar]
- Itch and nerve fibers with special reference to atopic dermatitis: Therapeutic implications. J Dermatol. 2014;41:205-12.
- [CrossRef] [PubMed] [Google Scholar]
- Increased nerve growth factor and its receptors in atopic dermatitis: An immunohistochemical study. Arch Dermatol Res. 2006;298:31-7.
- [CrossRef] [PubMed] [Google Scholar]




